
《GeoSeqBuilder》
引言
论文的引言首先概述了蛋白质序列-结构-功能关系的重要性,特别是蛋白质通过折叠形成特定的三级结构来执行其功能的过程。理解这种关系对于蛋白质结构预测和设计至关重要。近年来,得益于深度学习技术的进步和大量蛋白质序列数据的可用性,蛋白质结构预测取得了显著的进展。
传统的蛋白质序列设计方法依赖于物理能量函数或统计势函数,通过寻找能够以最低能量折叠成目标结构的序列。这些方法通常需要大量计算资源,并且实验验证的成功率较低,使得其在计算和实验方面的成本都非常高。因此,AlphaFold2等深度学习模型的出现引起了广泛关注。
尽管已有多种基于深度学习的蛋白质设计方法被开发出来,但大多数方法主要集中于提高序列恢复率,而较少进行实验验证。为了解决现有方法的局限性,作者提出了GeoSeqBuilder,这是一个基于几何特征和三体相互作用的深度学习框架,以提高设计效率和准确性。
GeoSeqBuilder 显式地对残基之间的三体相互作用进行建模,允许并行训练每个残基位置,从而避免了标签泄露,并加速了设计过程中的推理。通过在CATH 数据集上的测试,GeoSeqBuilder 实现了较高的残基恢复率,同时准确预测了侧链构象。此外,GeoSeqBuilder 生成的序列在实验验证中表现出稳定性和功能性。
总的来说,引言部分清晰地阐述了研究背景,讨论了当前方法的局限性,突出了深度学习在蛋白质设计中的重要性,并介绍了GeoSeqBuilder的创新点和潜在应用。
结果和讨论
Overview of the model architecture
GeoSeqBuilder 使用多尺度图卷积网络(GCN)来处理蛋白质的几何结构数据,模型通过以下步骤读取和学习数据:
1. 输入数据格式
GeoSeqBuilder 的输入数据主要是蛋白质的骨架结构,即由主链坐标(如 Cα 原子位置)构成的三维空间几何信息。模型将每个残基视为图中的节点,残基之间的几何相互作用被表示为图中的边(edges)。
数据特征包括:
- 残基坐标:表示每个残基在三维空间中的位置。
- 边缘特征:残基之间的距离、角度等几何信息。
2. 多尺度图卷积网络
模型采用多尺度的图卷积网络(GCN)来学习不同尺度的相互作用,步骤如下:
- 节点特征提取:从蛋白质骨架的几何特征中提取每个残基的局部环境信息,如相对位置、距离、角度等。
- 图卷积操作:通过邻近残基的特征来更新当前残基的特征。随着网络层数加深,残基能够获取更大范围的上下文信息。
- 1级网络:学习两体相互作用(如两个残基之间的距离)。
- 2级及更高级网络:学习三体及更高阶相互作用,捕捉更复杂的残基组合之间的关系。
3. 并行训练
GeoSeqBuilder 通过并行方式处理每个残基的预测任务。每个残基在不同尺度的图网络中独立学习其周围的几何环境特征,以便高效地推断残基的类型和侧链构象。
- 避免标签泄露:GeoSeqBuilder 使用了自掩码机制,确保模型仅使用当前和之前的已知信息,不会提前获取预测标签,避免“作弊”。
4. 训练目标
GeoSeqBuilder 通过最大化似然函数进行训练,生成与给定蛋白质骨架结构相匹配的残基序列。目标是最小化 -logP(Y|X),确保生成的序列与骨架相容。
5. 输出结果
经过学习过程,GeoSeqBuilder 最终能够生成适合给定蛋白质骨架的残基序列,并预测每个残基的侧链扭转角度。这些结果可以进一步用于实验验证,以评估生成的蛋白质序列是否能够正确折叠和保持功能性。
图解说明:GeoSeqBuilder 模型架构
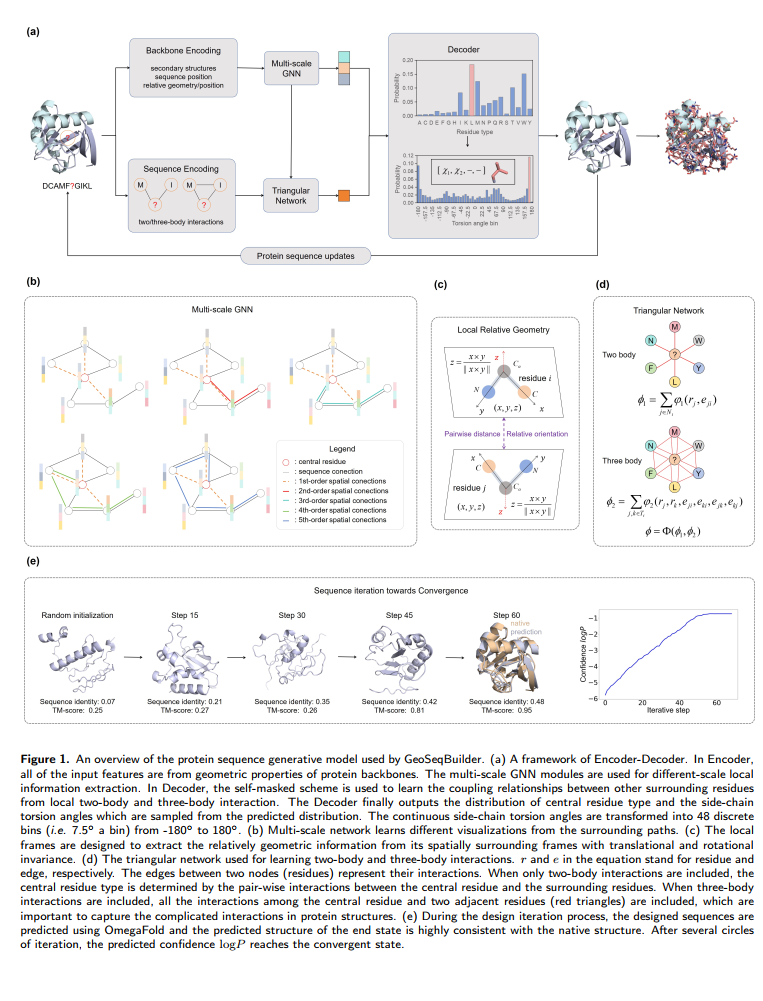
这张图展示了 GeoSeqBuilder 模型的整体架构和工作流程,主要用于基于几何深度学习的蛋白质序列生成。以下是对图中各部分的详细解释:
(a) Encoder-Decoder 框架
- Backbone Encoding(骨架编码):
- 提取蛋白质骨架中的特征,包括二级结构、序列位置和相对几何位置。这些特征被输入到多尺度图卷积网络(GNN)中。
- Sequence Encoding(序列编码):
- 通过捕捉蛋白质中的两体和三体相互作用,模型提取复杂的局部几何信息。
- Multi-scale GNN(多尺度图卷积网络):
- 提取不同尺度下的局部信息,帮助模型理解蛋白质残基之间的复杂相互作用。
- Decoder(解码器):
- 使用自掩码机制学习残基之间的耦合关系。解码器输出中心残基类型的分布和侧链的扭转角度,角度被离散化为 48 个区间。
(b) Multi-scale GNN(多尺度图卷积网络)
该模块显示了如何通过不同尺度(如一级、二级和更高阶)来连接节点和残基,从而捕捉不同空间层次的信息。每个节点代表一个残基,边表示残基之间的空间连接。
(c) Local Relative Geometry(局部相对几何)
展示了如何通过局部框架提取蛋白质几何信息,并保证旋转和平移不变性。通过计算残基之间的相对距离和方向来学习局部几何关系。
(d) Triangular Network(三体相互作用网络)
解释了如何通过三体相互作用预测中心残基类型。相比于仅考虑两体相互作用,三体相互作用网络还包含与相邻第三个残基形成的三角形相互作用,用于更好地捕捉复杂的蛋白质结构。
(e) 序列收敛过程
该部分展示了模型生成序列的迭代过程,以及如何通过 OmegaFold 验证生成的蛋白质结构与目标结构的匹配度。右侧的曲线展示了随着迭代次数的增加,生成序列的置信度 logP逐渐收敛。不同迭代步骤展示了模型从随机初始化到最终收敛时的 TM-score(结构相似性评分)和序列相似性。
总结
GeoSeqBuilder 通过多尺度图卷积网络捕捉蛋白质局部的几何信息,结合三体相互作用网络,生成与目标骨架结构高度一致的蛋白质序列。该模型能够有效地处理蛋白质残基之间复杂的相互作用,并在生成过程中通过反复迭代保证结果的准确性和一致性。
蛋白质侧链构象预测分析
在 GeoSeqBuilder 模型中,侧链构象的预测依赖于解码器模块,结合几何特征、残基类型以及局部相互作用信息生成侧链扭转角。以下是对蛋白质侧链构象预测部分的详细分析:
1. 侧链扭转角度的离散化处理
GeoSeqBuilder 将侧链的二面角(如 (_1)、(_2) 等)划分为 48 个离散区间(bin),每个区间覆盖 -180° 到 180° 的角度范围。这使得模型能够以概率分布的方式选择每个残基的侧链二面角。
2. 预测准确性
通过最大化侧链二面角的似然函数,GeoSeqBuilder 为每个残基选择最适合其几何结构的扭转角。模型在核心区域的疏水残基预测表现出高精度,而在暴露于溶剂中的残基,由于其更高的灵活性,预测的准确性稍低。
3. 结构依赖性
GeoSeqBuilder 充分利用了蛋白质的局部几何信息(如残基间的相对位置、距离和角度),这些信息在侧链构象预测中至关重要。通过三体相互作用网络,模型捕捉了残基的复杂几何约束,从而提高了侧链的预测质量。
4. 侧链构象与二级结构的关系
根据残基所在的二级结构类型,模型的预测准确度不同: - 螺旋结构:螺旋中的残基扭转角相对稳定,预测准确度较高。 - β折叠:疏水残基在 β 折叠中通常埋藏在核心区域,预测准确度较高。 - 环结构:环结构中的残基由于更高的灵活性,预测难度较大,特别是暴露在溶剂中的残基。
5. 氢键和其他相互作用
氢键等弱相互作用对侧链构象预测的影响较大,尤其是极性和带电残基。GeoSeqBuilder 在处理这些残基时,额外考虑了几何约束,以确保氢键的形成。
6. 置信度评估
通过计算侧链构象的熵值,模型评估了预测的置信度。对于蛋白质核心区域的残基,预测置信度较高,因为这些区域的构象较为保守。暴露于溶剂中的残基由于其灵活性较高,置信度较低。置信度分布通常呈现U 形曲线,表明在高保守性和高灵活性区域,置信度有所不同。
总结
GeoSeqBuilder 通过离散化侧链扭转角并结合三体相互作用网络,成功捕捉了蛋白质结构中的几何约束和相互作用。模型在预测疏水核心区域和螺旋结构中表现出较高的准确性,但在处理暴露于溶剂中的灵活残基时,预测精度相对较低。
Discussion 部分详述
GeoSeqBuilder 的讨论部分通常涉及对研究结果的解释、方法的优势与创新、局限性以及未来工作方向。以下是可能的讨论内容结构:
1. 结果总结与解读
作者首先总结了 GeoSeqBuilder 的关键结果,并对其进行了深入解释: - 模型在蛋白质序列恢复和侧链构象预测中表现出色,使用几何深度学习和多尺度图卷积网络显著提高了预测精度。 - 模型的序列恢复率在低丰度残基的预测上也表现优异,表明 GeoSeqBuilder 在处理极端条件时具有鲁棒性。
2. 方法的优势与创新
在这部分,讨论了 GeoSeqBuilder 的创新性以及相对于现有方法的优势: - 几何特征建模的优势:通过使用局部几何框架和三体相互作用,GeoSeqBuilder 能够捕捉蛋白质中复杂的相互作用,而不仅限于简单的两体相互作用。 - 自掩码机制:相比传统的自回归模型,自掩码机制使得序列生成过程中更加精准,避免了标签泄露。 - 多尺度图卷积网络:模型能够捕捉到蛋白质结构中局部和全局相互作用的平衡关系,有助于提升预测效果。
3. 模型局限性
讨论部分指出了 GeoSeqBuilder 的局限性: - 计算复杂度:尽管 GeoSeqBuilder 提供了高精度预测,但它在计算资源上要求较高,尤其是在处理大规模蛋白质结构时。 - 实验验证不足:模型尚未在广泛的蛋白质结构上进行实验验证,进一步的实验研究是必要的。 - 动态蛋白质的预测:模型基于静态结构数据,未考虑蛋白质的动态特性,可能影响处理柔性蛋白时的预测能力。
4. 与现有工作的比较
在与其他方法的对比中,GeoSeqBuilder 展现了其独特的优势: - 与基于能量函数或其他深度学习方法相比,GeoSeqBuilder 在捕捉残基间复杂的三体相互作用时表现更好。 - 相比 ProteinMPNN,GeoSeqBuilder 在低丰度残基的预测上表现更加准确。
5. 未来工作方向
作者提出了未来研究和改进的方向: - 改进计算效率:未来可能会优化模型的计算性能,使其能够处理更大规模的蛋白质结构。 - 动态蛋白质建模:将动态特性纳入模型,使其能够预测柔性和多构象蛋白质的行为,是未来的研究方向之一。 - 更多实验验证:扩展实验验证范围,进一步评估 GeoSeqBuilder 在更多蛋白质结构上的适用性。 - 新功能蛋白设计:研究 GeoSeqBuilder 在设计新型功能蛋白中的潜力,特别是在生物医药领域的应用。
结论
讨论部分强调了 GeoSeqBuilder 在蛋白质序列设计中的创新贡献,尤其是在捕捉几何相互作用和提高序列恢复率方面。同时,作者也诚实地评估了模型的局限性,并提出了未来可能的改进和研究方向。


